3月2日,國際心臟研究學會官方期刊Journal of Molecular and Cellular Cardiology在線發表了中國科學院上海營養與健康研究所楊黃恬研究員與香港城市大學尹慧勇教授合作的研究論文“Uncoupling protein 3 protects against pathological cardiac hypertrophy via downregulation of aspartate”。該研究揭示線粒體解偶聯蛋白3(UCP3)通過調控天冬氨酸代謝抑制病理性心肌肥厚的作用和分子機制,為靶向線粒體代謝重塑治療心肌肥厚提供了新的理論依據和潛在干預策略。
病理性心肌肥厚是高血壓、主動脈狹窄等壓力超負荷性心臟疾病的重要病理特征,伴隨著線粒體蛋白異常表達,代謝重塑及三羧酸循環中間產物的異常變化。然而,在心肌肥厚過程中,線粒體膜蛋白調控三羧酸循環中間產物的分子機制尚未明確。UCP3是線粒體內膜的陰離子轉運體,既往楊黃恬研究組發現其通過與線粒體通透性轉換孔調控蛋白結合保護心肌細胞對抗缺血損傷,但其在壓力超負荷性心肌肥厚中的作用和機制尚不清楚。
研究團隊發現,在主動脈縮窄術(TAC)誘導的小鼠壓力超負荷病理性心肌肥厚模型及去甲腎上腺素誘導的新生大鼠心肌細胞中,UCP3表達顯著下調。通過分別構建全身性及心肌細胞特異性UCP3敲除小鼠和心肌細胞特異性UCP3過表達小鼠,研究人員發現TAC誘導的心肌肥厚與心功能障礙在UCP3缺失小鼠中顯著加重,而UCP3過表達則能有效改善上述病理表型。進一步在細胞實驗中證實,UCP3敲低加重去甲腎上腺素誘導的心肌細胞肥大,而過表達UCP3則抑制這一過程。
機制研究發現在TAC誘導的小鼠心肌肥厚模型中,谷草轉氨酶2(GOT2)活性及天冬氨酸水平顯著升高,而UCP3敲除加重這一現象,過表達UCP3則逆轉其異常。在去甲腎上腺素誘導的細胞中,UCP3敲低進一步升高GOT2活性,而UCP3過表達則減弱其升高,同時伴有天冬氨酸水平的下調。值得注意的是,去甲腎上腺素刺激削弱了UCP3與GOT2的內源性結合,而外源性補充天冬氨酸可抵消UCP3過表達對心肌細胞肥大的保護作用,提示UCP3抑制GOT2活性,降低天冬氨酸積累是其抗心肌肥大的關鍵機制。
該研究揭示了UCP3在壓力超負荷性心肌肥厚中的保護作用,并表明了天冬氨酸在UCP3調控心肌肥厚中的核心作用,為開發基于代謝干預的心肌肥厚治療策略提供了重要科學依據。
中國科學院上海營養與健康研究所楊黃恬研究員、香港城市大學尹慧勇教授為本論文共同通訊作者,中國科學院上海營養與健康研究所博士畢業生王亞軍、助理研究員譚吉良博士、博士畢業生李露曉為該論文的共同第一作者。研究獲得國家自然科學基金和香港研究資助局的資助以及中國科學院上海營養與健康研究所所級公共技術中心分析測試技術平臺和實驗動物技術平臺的支持。
文章鏈接:
https://www.sciencedirect.com/science/article/abs/pii/S0022282825000410
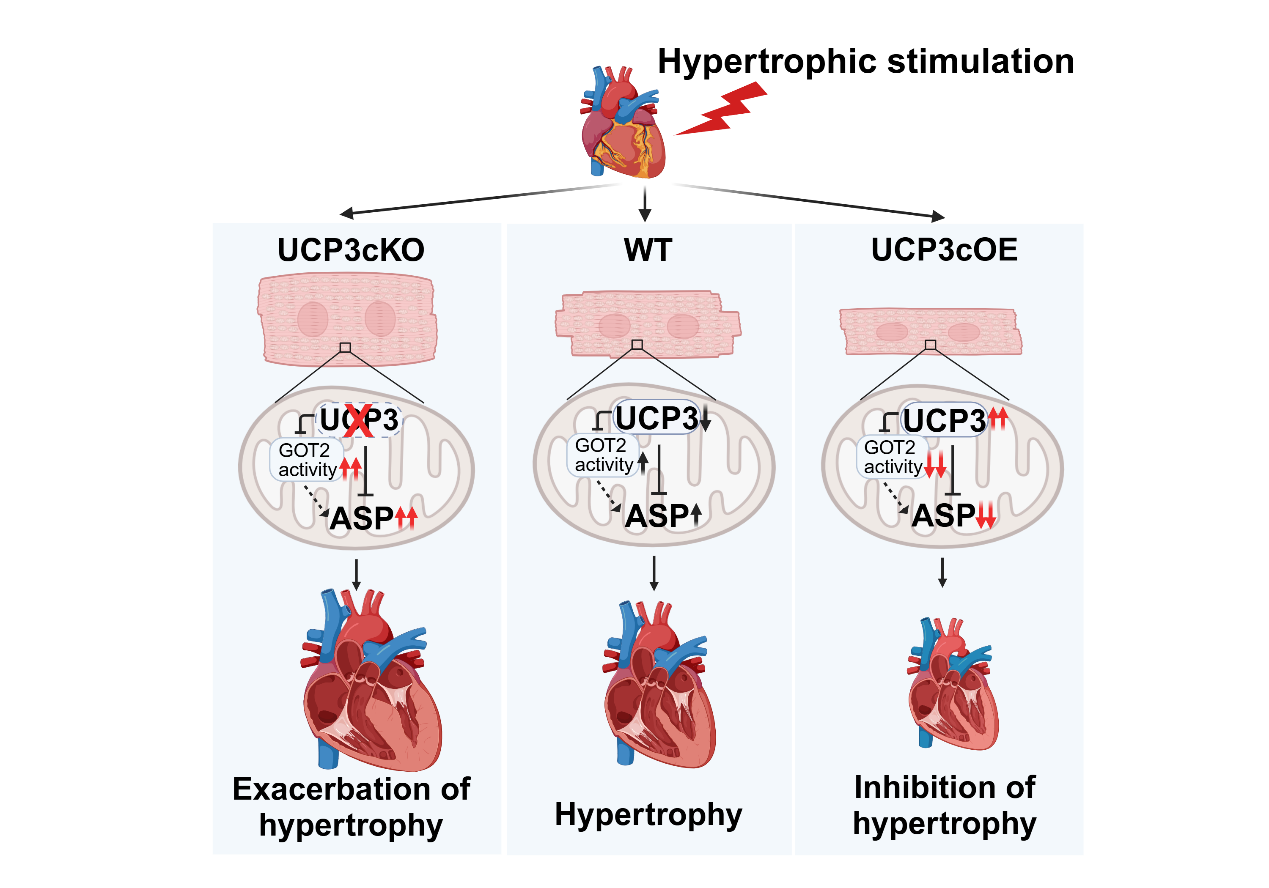
圖:UCP3通過抑制天冬氨酸累積發揮抵抗心肌肥大的作用
 官方微信
官方微信